
Publication dans J. Phys. Chem. B
Equipe LMCT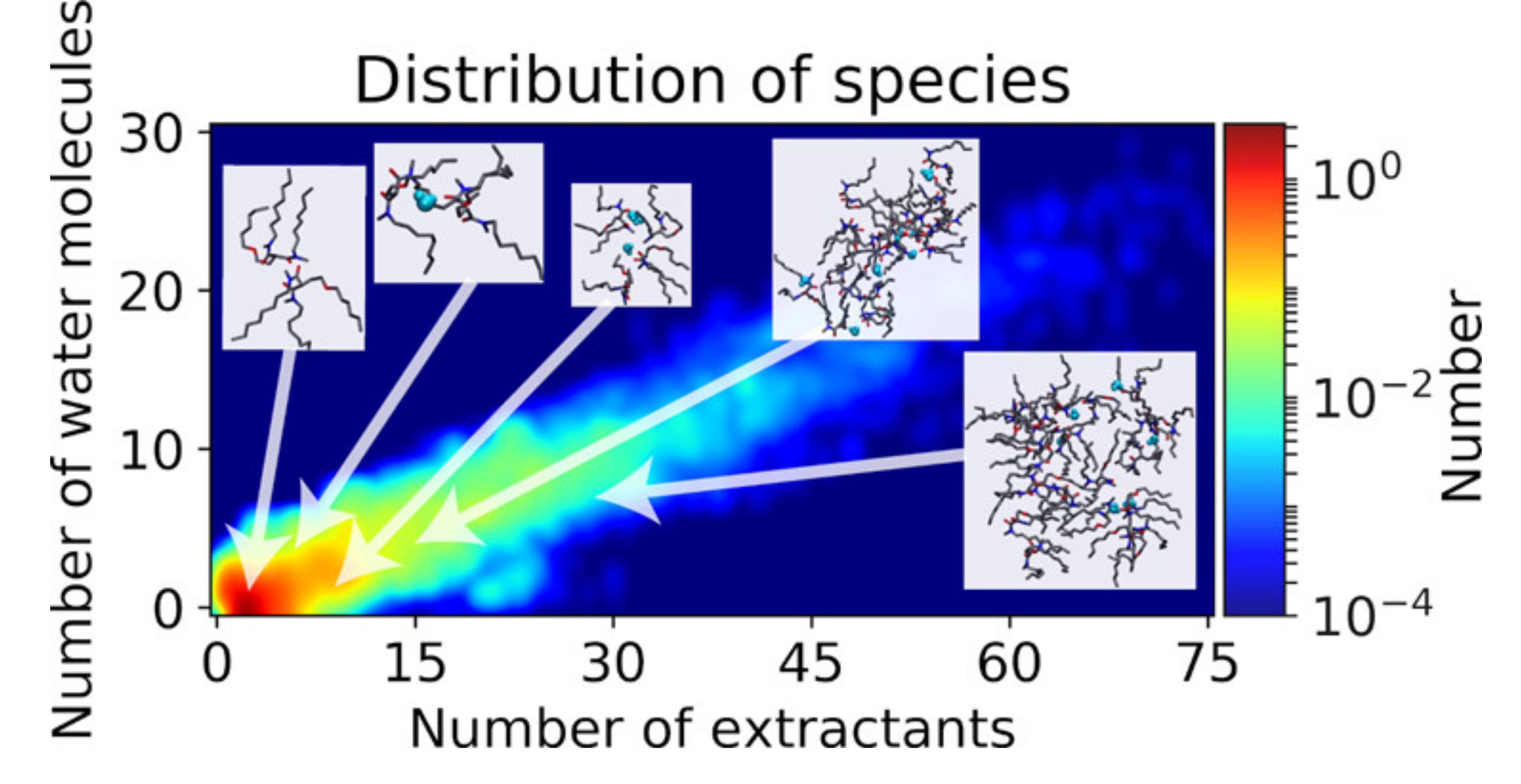
The aggregation of malonamide extractants diluted in an aliphatic solvent phase has been studied in the presence of water by molecular dynamics simulation. Using association criteria based on distances between molecules and graphs theory, the aggregate distribution has been computed and the corresponding Gibbs energy of aggregates and mass action law constants have been determined. Finally, a model allowing us to the compute critical micelle concentration and osmotic data for a variable concentration of extractants, with or without a correction of the organic phase activity, was developed. It appears however that the accurate depiction of the aggregation allows modeling the thermodynamics of the solution even without an explicit calculation of the activity: both models give results in good agreement with the experiments.
Pour plus d'information, lire l'article de Marin Vatin, Magali Duvail, Philippe Guilbaud et Jean-François Dufrêche dans J. Phys. Chem. B, 2021, 125, 3409
![]()
![]()
![]()
![]()
![]()
![]()

![]()

![]()