
Chercheure CEA/DRF
Ph.D. Chimie (HDR)
ICSM/LMCT (Bât. 426)
Site de Marcoule
BP 17171
F-30207 Bagnols-sur-Cèze Cedex
Tel : 04 66 79 57 21
Fax : 04 66 79 76 11
e-mail : magali.duvail ad cea.fr

Collaborations : Pr. Th. Zemb (ICSM/LTSM), Pr. L. Arleth (University of Copenhagen, Danemark), Dr. S. Marcelja (Australian National University, Canberra)
La modélisation mésoscopique des propriétés thermodynamiques des microémulsions (mélange eau / huile / surfactant) est un enjeu fondamental à la compréhension des phénomènes mis en jeu lors de l’extraction liquide-liquide, utilisée pour la séparation poussée des ions. Les objectifs principaux de cette étude sont de :

Extrait du film sur la modélisation mésoscopique (lien YouTube)
Collaborations : Dr. Ph. Guilbaud (CEA), Pr. J.-F. Dufrêche (ICSM), Pr. M. Jardat (PHENIX, Sorbonne Université)
Des approches par modélisation multi-échelle sont développées et mises en oeuvre pour determiner les propriétés thermodynamiques des ions en solution, notamment les sels de lanthanides (Ln3+) et d'uranyle (UO22+). Ces approches couplent des simulations de dynamique moléculaire classique avec polarisation explicite et des simulations « gros-grains ». Par exemple, par le calcul des potentiels de force moyenne, nous pouvons analyser les processus d’association / dissociation des complexes formés en solution.
Nous pouvons également accéder aux coefficients d'activités des électrolytes en solution, directement comparables aux expériences, par modélisation moléculaire de l'équilibre osmotique.
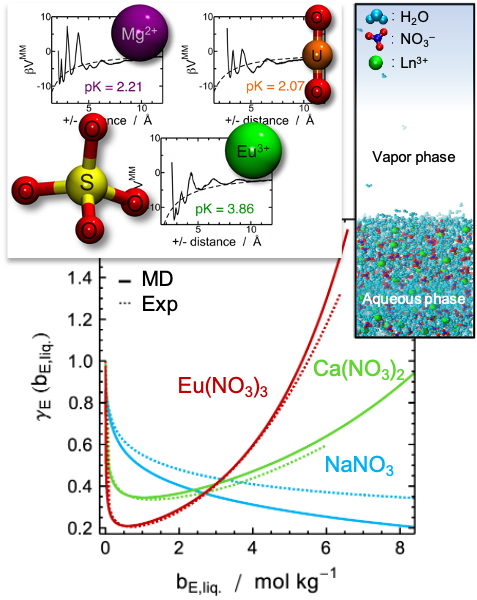
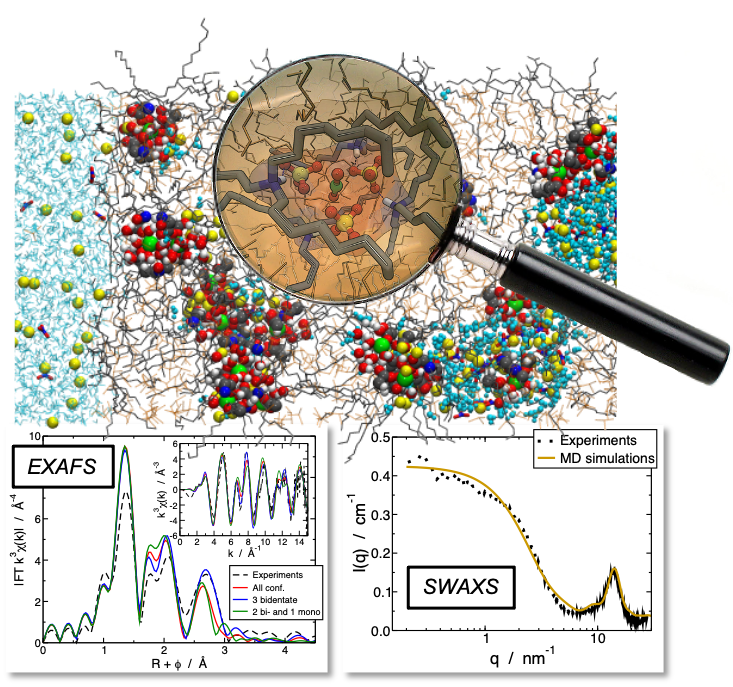
Des simulations de dynamique moléculaire sur la solvatation de lanthanides en phase organique (alcool et alcane) indiquent que ces simulations peuvent considérablement améliorer les connaissances sur la solvatation des éléments f en phase organique. Nous nous intéressons également aux propriétés thermodynamiques de micelles composées d'ions, d'eau et de molécules extractantes dans des solvants apolaires, et plus particulièrement à leurs énergies libres de courbure. Ces simulations ouvrent la voie à d’autres études structurales dans les solvants organiques (coordination des anions, complexation par des ligands monoamide, diamide ou azotés), et donc à des progrès en ce qui concerne la spéciation en phase organique.
Collaborations : Dr. S. Dourdain (ICSM/LTSM), S. Pellet-Rostaing (ICSM/LTSM), Dr. S. Le Crom (ICSM/LMCT)
Projet : Lab'UM Chimie (Université de Montpellier) RAMELI
L'objectif du projet RAMELI (Rationalisation par dynamique moléculaire des effets de structure dans les Mécanismes d'Extraction en milieu Liquide Ionique) consiste à étudier et comprendre l'organisation structurale des phases d'extraction en milieu liquide ionique, afin de comprendre et de prédire leurs propriétés d'extraction. Ce projet s’appuie sur des résultats obtenus dans le cadre d’études menées à l’ICSM pour le développement de procédés d’extraction en milieu liquide ionique qui ont permis de démontrer que les meilleures performances d’extraction en milieu liquide ionique ne sont pas uniquement dues à des mécanismes de complexation différents, mais à des propriétés structurales différentes.
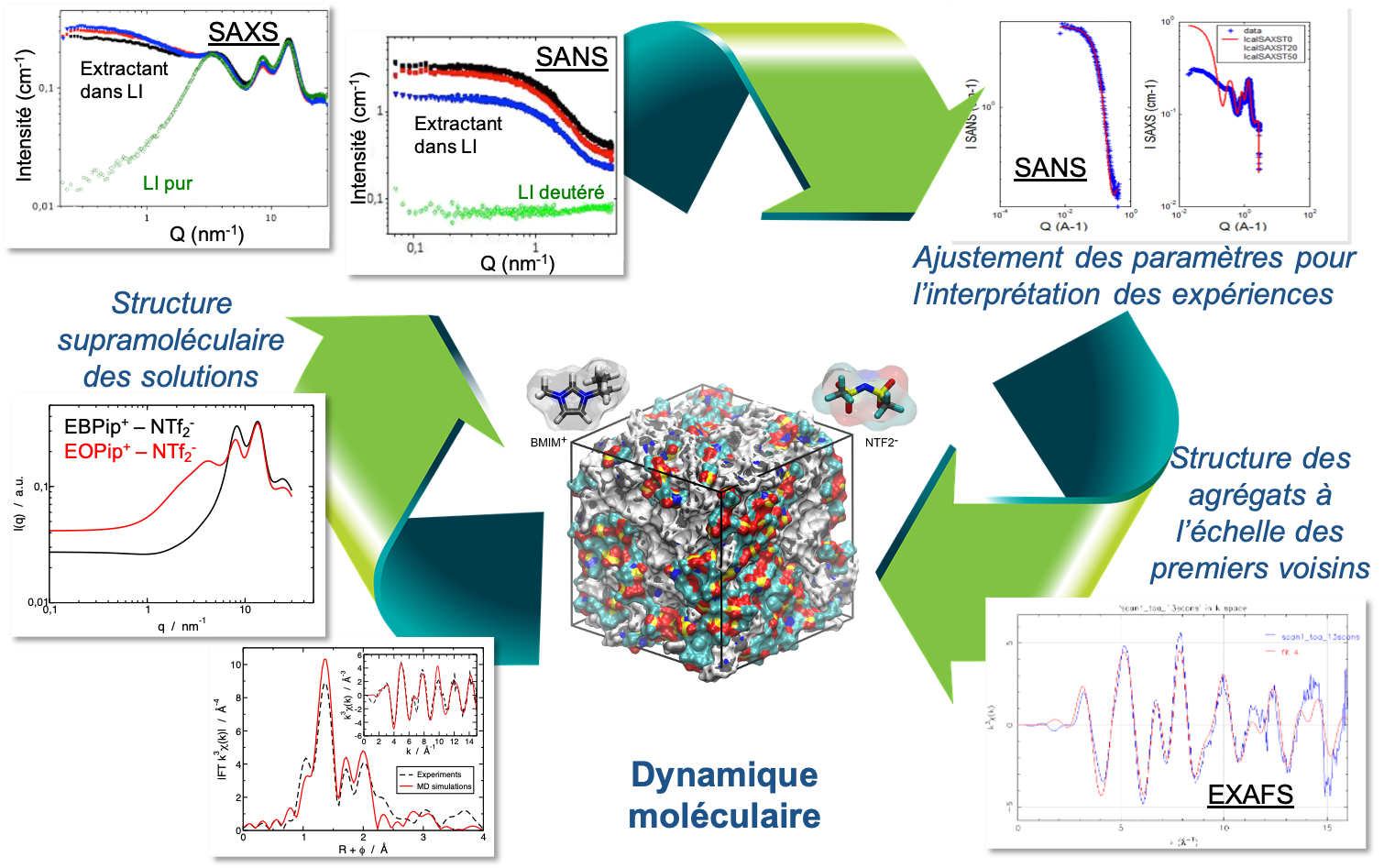
Aucune démarche expérimentale n’ayant permis à ce jour de décrire complétement l’organisation structurale des phases d’extraction en liquide ionique, ce projet a pour but de rationaliser tous les résultats expérimentaux en simulant ces phases par dynamique moléculaire. En permettant de définir des modèles d’ajustement des données précédemment acquises, cette approche théorique permet de décrire la structures multi-échelle de ces phases (complexes, agrégats, structures du liquide ionique) et ainsi d’apporter des éléments de compréhension cruciaux sur l’impact des effets structuraux sur les performances d’extraction.
Collaborations : Dr. A. Poulesquen (CEA), Dr. D. Petit (L2C, Université Montpellier), J. Lind (Woellner GmbH & Co.KG, Allemagne)
Projet : ANR DYNAMISTE ANR-15-CE07-0013
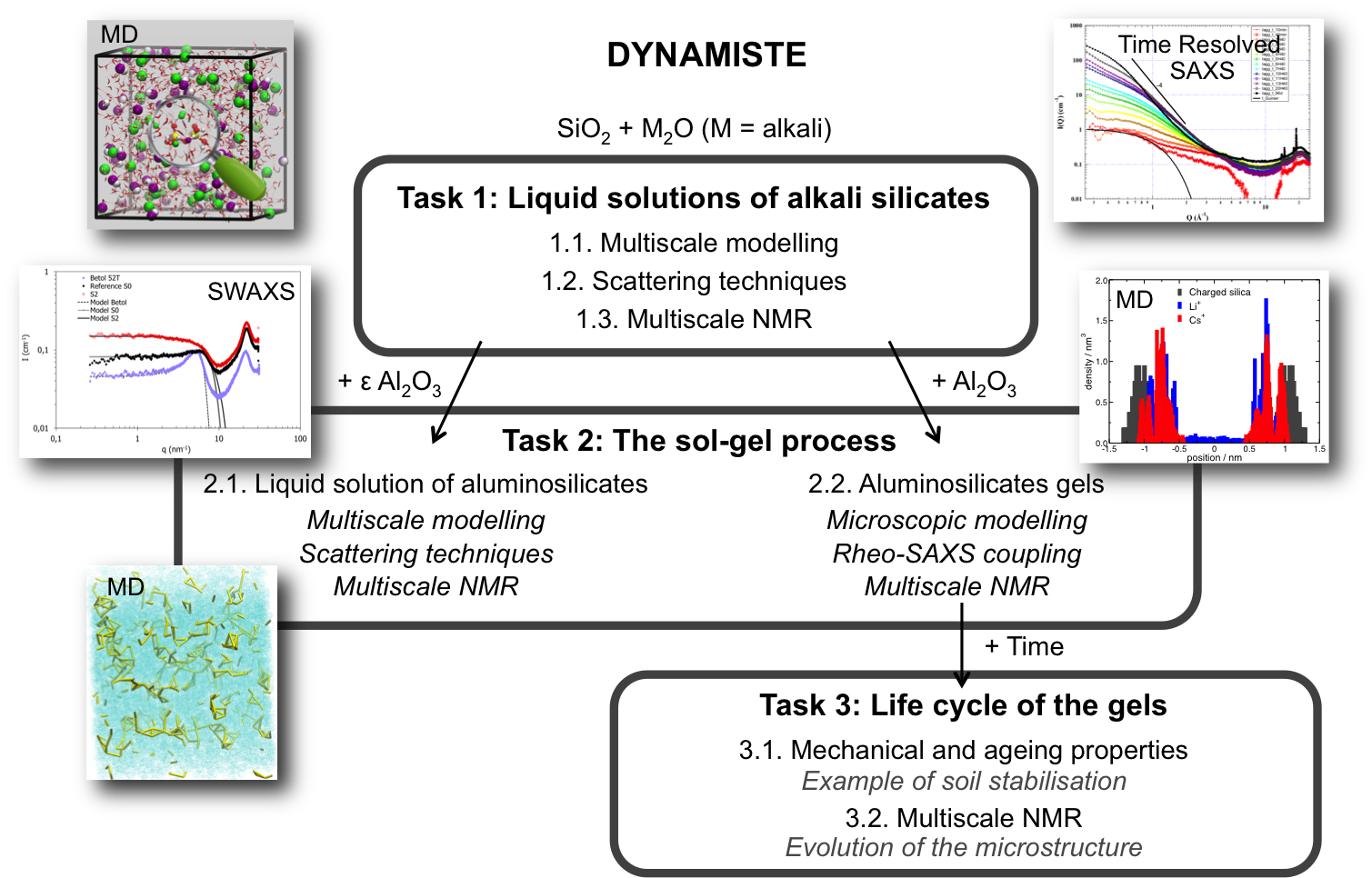
Le projet DYNAMISTE (DYNamique de fluides AluMIno-SilicaTES) a pour objectif le développement d’outils expérimentaux et théoriques afin d’optimiser les procédés industriels dans lesquels sont impliquées les solutions d’aluminosilicates, comme la liquéfaction de la céramique, les liants pour les peintures minérales, les mortiers réfractaires ou la stabilisation des sols, entre autres, dans une optique de développement d’industries propres. Il rassemble les compétences de l’ICSM, du Département de recherche sur les technologies pour l'Enrichissement, le Démantèlement et les Déchets du CEA Marcoule et le Laboratoire Charles Coulomb (Université Montpellier), en collaboration avec un partenaire industriel allemand, Wöllner GmbH & Co. KG, qui est un des leaders dans le domaine de la production de solutions alcalines silicatées.
Afin d’accéder à tous les phénomènes spatio temporels de ces systèmes, la partie expérimentale de ce projet repose sur (i) des études basées sur des techniques de rhéologie couplées à des techniques de diffusion : Dynamic light scattering (DLS), Small et Wide Angle x-ray Scattering et diffraction (SWAXS et XRD) et (ii) une approche multi-échelle par RMN (de l’Å à quelques dizaines de µm). Dans le même temps, la partie théorique est basée sur une approche multi-échelle couplant des simulations de dynamique moléculaire (au niveau microscopique) à des simulations gros grains (à l’échelle mésoscopique), ce qui permet d’accéder aux propriétés structurales et dynamiques de ces fluides.
![]()
![]()
![]()
![]()

![]()


![]()
 English version
English version orcid.org/0000-0003-1586-260X
orcid.org/0000-0003-1586-260X