
New results (14/05/2025)
Team LMCTThe surface of uranium dioxide (UO2) plays a central role in the dissolution, reprocessing and long-term storage of spent fuel. To better characterise this interface under aqueous conditions, researchers at the ICSM used ab initio molecular dynamics (DFT-MD) simulations, capable of following the evolution of water molecules interacting with the three most stable crystallographic surfaces of UO2: (111), (110) and (100).
This work highlights very different adsorption behaviours depending on the surface: surface (100), which is particularly reactive, leads to complete and rapid hydroxylation, synonymous with passivation. Conversely, surfaces (111) and (110) favour mainly molecular adsorption, leading to the formation of structured layers of water. The simulations also reveal a clear electrostatic organisation, reflected by the emergence of differentiated surface charges.
These results make it possible to map, with atomic resolution, the state of hydration of UO2 surfaces in equilibrium with water. They provide key insights into the early stages of interfacial reactions involved in fuel cycle processes.
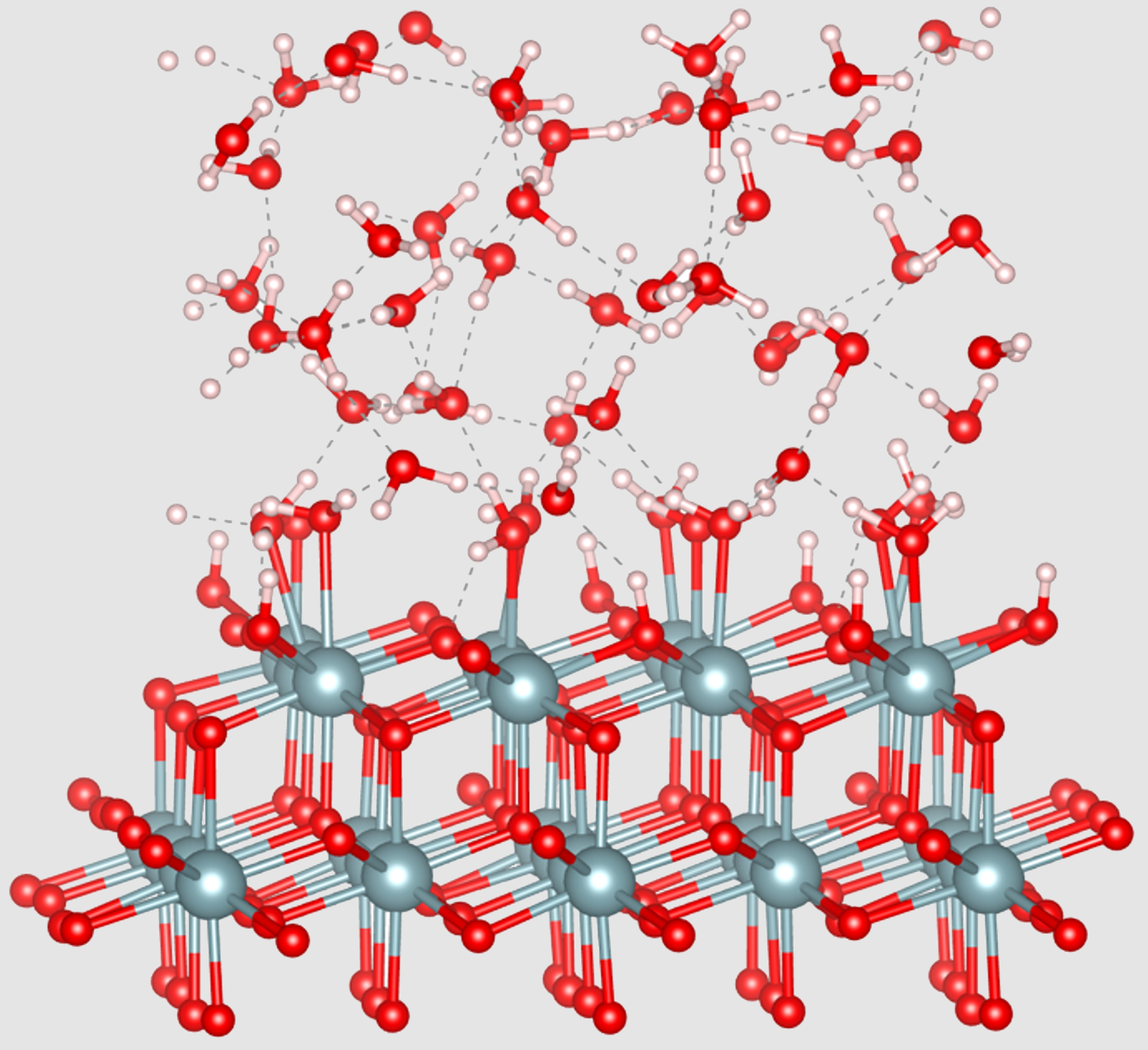
Credit: ICSM/LMCT
For more information:
Structure and chemistry of uranium dioxide (UO2) in water from first-principles molecular dynamics simulations
G. Rodrik, Y. Foucaud, B. Siboulet, M. Duvail, S. Szenknect, J.-F. Dufrêche. Colloids and Surfaces A: Physicochemical and Engineering Aspects 721 (2025), 136827. DOI: 10.1016/j.colsurfa.2025.136827
![]()
![]()
![]()
![]()
![]()
![]()

![]()

![]()