
Nouveaux résultats (14/05/2025)
Equipe LMCTLa surface du dioxyde d’uranium (UO2) joue un rôle central dans les processus de dissolution, de retraitement ou encore de stockage à long terme des combustibles usés. Pour mieux caractériser cette interface en conditions aqueuses, des chercheurs de l’ICSM ont eu recours à des simulations de dynamique moléculaire ab initio (DFT-MD), capables de suivre l’évolution des molécules d’eau en interaction avec les trois surfaces cristallographiques les plus stables de l’UO2 : (111), (110) et (100).
Ces travaux mettent en évidence des comportements d’adsorption très contrastés selon la surface : la face (100), particulièrement réactive, conduit à une hydroxylation complète et rapide, synonyme de passivation. À l’inverse, les surfaces (111) et (110) favorisent une adsorption principalement moléculaire, donnant lieu à la formation de couches structurées d’eau. Les simulations révèlent également une organisation électrostatique nette, traduite par l’émergence de charges de surface différenciées.
Ces résultats permettent ainsi de cartographier, avec une résolution atomique, l’état d’hydratation des surfaces d’UO2 en équilibre avec l’eau. Ils apportent des éléments clés pour éclairer les premiers stades des réactions interfaciales impliquées dans les procédés du cycle du combustible.
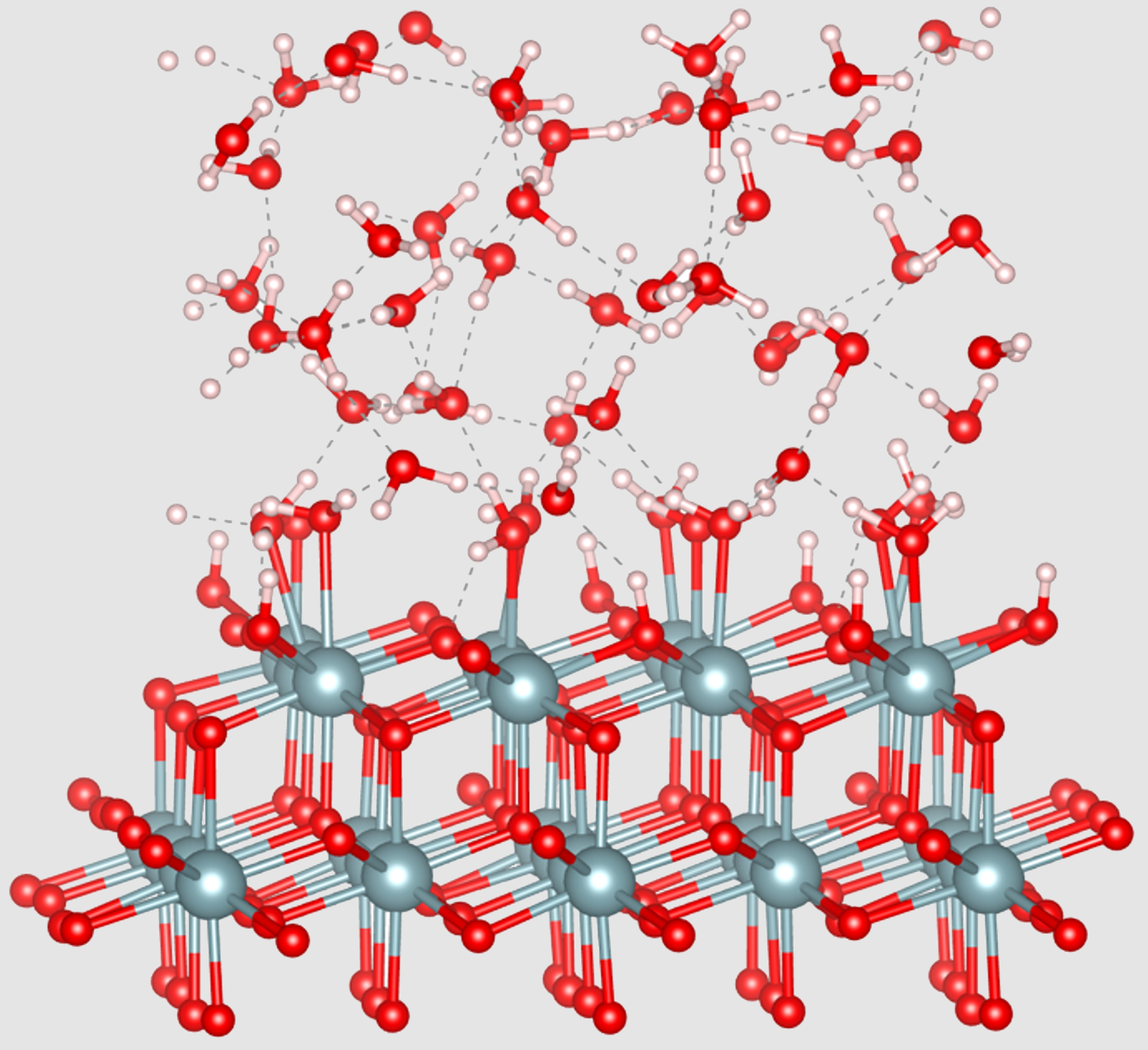
Crédit : ICSM/LMCT
Pour plus d’information :
Structure and chemistry of uranium dioxide (UO2) in water from first-principles molecular dynamics simulations
G. Rodrik, Y. Foucaud, B. Siboulet, M. Duvail, S. Szenknect, J.-F. Dufrêche. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 721 (2025), 136827. DOI: 10.1016/j.colsurfa.2025.136827
![]()
![]()
![]()
![]()
![]()
![]()

![]()

![]()